The hype on AlphaFold keeps growing with this new preprint
Original Source Here
This story is based on a preprint just posted in the bioRxiv that formally describes a tool dubbed ColabFold under the moto Making protein folding accessible to all (which I would have rather phrased Making modern protein structure modeling accessible to all).
Created by Milot Mirdita from the Max Planck Institute for Biophysical Chemistry in Germany, Sergey Ovchinnikov from Harvard University in the U.S. and Martin Steinegger from the Seoul National University in South Korea, ColabFold is a set of Google Colab notebooks evolved from the early prototypes, that allow users having nothing more than a computer, internet connection and a free Google account, to run protein structure predictions using the latest cutting-edge machine learning technologies on hardware provided by Google; in addition benefiting from several optimizations that reduce running time without compromising the quality of the results and capitalizing on a modern tool for the fast generation of multiple protein sequence alignments -which as I explained in other stories are important to ensure accurate results.
More precisely, ColabFold allows users to run AlphaFold2 or RoseTTAFold (an academic AI-based program -from the Baker lab, one of the academic leaders in protein structure prediction- that came up after CASP14 so it hasn’t yet been formally evaluated, but apparently performs close to AlphaFold2) combined with fast multiple sequence alignment generation by the program MMseqs2. Users can upload their own alignments too, which may be handy for very tricky proteins or protein families, or for alignments coming from proprietary data such as metagenomics projects.
Optimized generation of protein sequence alignments with MMseqs2 improves models and reduces running time
As shown widely in the CASP papers of the recent years (see for example my peer-reviewed CASP13 assessment), it is key for the performance of all these methods to count with vast multiple sequence alignments, rich in numbers of sequences that, in the ideal case, cover the whole target protein smoothly. The initial compilation of the alignment is thus critical. Further complicating this, typical databases of protein sequences contain millions to billions of sequences, but of course only very small subsets of them correspond to proteins of the same structural family which are the ones that one wants to show up in the multiple sequence alignment. That’s where the MMseqs2 component of ColabFold comes in.
MMseqs2 by Martin Steinegger and Johannes Söding is a program for sensitive protein sequence searches inside huge sequence dabatases. I will not go into any detail but the paper describing MMSeqs2 might be of interest to data scientists as the core aim of the program is to accelerate sequence searches. ColabFold executes the MMseqs2 program through API calls to a dedicated server. The authors optimized the size and variability of the protein sequences contained in their sequence databases so that by running the program iteratively a few times they can produce ample, informative yet tractable alignments. In the preprint the authors in fact show that these multiple sequence alignments produced by MMseqs2 lead AlphaFold 2 to more accurate predictions than those it obtains through its custom multiple sequence alignments, and runs around one order of magnitude faster.
Beyond simple modeling of monomeric “isolated” proteins
A vast majority of proteins do not work as isolated molecules but rather as complexes, either with themselves (so-called homo-dimers, homo-trimers, and so on, or homooligomers in general) or with other proteins (called hetero-dimer, hetero-trimer, etc.). The main evaluation were AlphaFold2 was found to “win” in CASP14 was in modeling the structures of proteins by themselves, but there was also some indication that AlphaFold2 was correctly modeling protein complexes too. This was further explored in the early Google Colab notebooks by Minkyung Baek and Yoshitaka Moriwaki, and then by the authors of ColabFold who ended up integrating this possibility into the released notebooks. Thus, ColabFold users can easily model isolated proteins as well as their homo- and hetero-complexes. For heterocomplexes it is tricky to develop alignments, but the ColabFold authors already dealt with all this burden making a very simple interface for the users to simply tick for oligomerization states and enter the different sequences of the proteins involved.
Notably, AlphaFold has no way to tell by itself of a protein is monomeric, homodimeric, heterodimeric with another protein, etc. This information is sometimes known from biochemical or biophysical experiments, in which case it is used as an input. If no hint is available about this, then users should probably run the predictions in different modes and compare the results critically.
Flavors for lay and advanced users
As described in the official GitHub page here, ColabFold includes various notebooks tailored for different kinds of runs: one for RoseTTAFold, one for AlphaFold2 in a simple mode that allows running only monomeric proteins but with minimal decisions to be made, and another for AlphaFold2 with fully exposed features which allows the full control of oligomerization states among other options that users can experiment with.
Depending on the exact GPU resources allocated when the users logs into Google Colab, it is possible to model proteins of as many as 1000 to 1400 amino acids, which covers a substantial number of the proteins of interest. For bigger proteins, or for more privacy or convenience, users can also get the generated code, download the whole AlphaFold program and MMseqs2-generated alignments, and run everything locally with its own GPUs (and even benefit from the code in the notebook).
Quality estimates
These user-friendly interfaces not only provide users with models of the protein structures, but also with estimates of their qualities. Such estimates are essential, I’d say as important as the predictions themselves, so they should rather be accurate. That’s because the end user should know what regions of the models are reliable, i.e. likely to be similar to the true structure, and which regions might not be well predicted. When I was an academic assessor during CASP13 (this academic paper) I stressed the importance of producing three kinds of quality estimates: one that measures the global quality of the overall fold, another that measures the quality of each amino acid individually, and another that measures the quality of the relative distances and orientations between any pair of amino acids in the protein. The ColabFold notebooks provides all three metrics, as I show in the example below.
Full online visualization of the results -an example run
All tunable variables are input in the fields of a quite rich GUI, on top of which users can of course modify the code by hand. But the cozy interface is not limited to the inputs. The notebooks feature rich graphical outputs with which users can inspect the run as it goes, and at the end inspect all the quality estimate plots and even the 3D models right inside the browser.
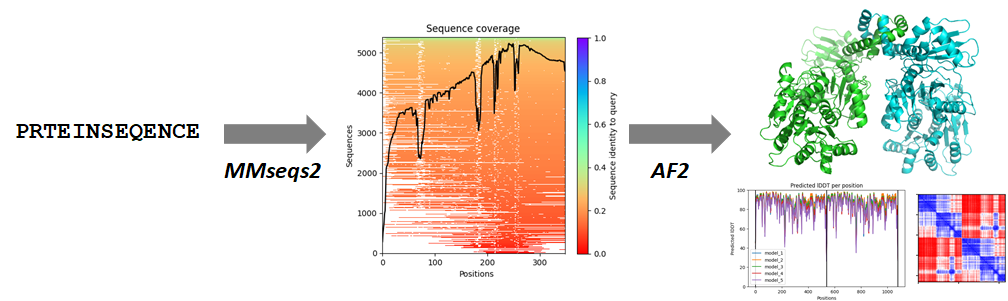
See these example outputs from an actual run I did on a protein that I know is an homodimer but whose structure I have very few clues about. After installing the required software with a first click in the ColabFold notebook, then setting up the protein’s sequence and telling the notebook that I know my protein is an homodimer, I first run the MMseqs2 module of the notebook to get a multiple sequence alignment that the notebook will feed into AlphaFold in the next step. The alignment returned by MMseqs2 is characterized by this summary graph, where we would like to have more green-cyan-bluish tones at the top and ideally a flatter black profile, although this doesn’t look too bad:
Once the alignment is done (ideally one should download and explore that its sequences are good enough, but this is hard because MMseqs2 returns very large numbers of hits) I move on to the next module of the notebook, where I start AlphaFold2. While it runs, the notebook allows me to monitor each of the 5 predicted models as they are produced:
At the end of the AlphaFold run I can inspect each of the 3D models right there thanks to a 3Dmol plugin. Notice that each of the 5 models comes out with an average predicted LDDT score, which is used to rank the models. In this case all 5 models are quite good all along the sequence (indicated by high pLDDT values), and they are all very similar to each other with quite high average pLDDT. The PAE plots do reveal however some uncertainty in the relative position of the two protein chains that make the dimer (shades of white to red in the Predicted Aligned Error plots):
Refinement of the models with molecular simulations
The models produced by AlphaFold or any other modeling method can have various problems such as clashes between atoms, unfulfilled interactions, etc. When models are predicted to be of high quality, it then makes sense to further improve them through molecular dynamics simulations. Briefly, in such simulations the protein models are described at full-atom level, sometimes even with simulated water molecules around, and allowed to fluctuate under realistic physics at a given temperature and pressure. The aim is to remove any unrealistic clashes, optimize geometries, and satisfy interactions and packing, especially of the amino acid side chains.
There are several programs that allow running molecular dynamics simulations. Users could download all models and run these simulations in their local computers with their own methods and pipelines. But ColabFold allows users to run the simulations right in the notebook, provided equilibrated versions of the models in-place.
Closing words
The coupling of MMseqs2 to AlphaFold2 within Google Colab provides free access to the state of the art of protein structure prediction, without the need of any specialized, expensive hardware, nor any software installs. As I discussed here, this is poised to revolutionize biology, and the best is that it is accessible to researchers all around the world, for free.
Links to literature, code and notebooks
The preprint at bioRxiv: https://www.biorxiv.org/content/10.1101/2021.08.15.456425v1.full.pdf
ColabFold GitHub, with links to all release and prototype notebooks: https://github.com/sokrypton/ColabFold
MMseqs2 in Github: https://github.com/soedinglab/MMseqs2
MMseqs2 paper: https://www.nature.com/articles/nbt.3988
Previous stories by me on AlphaFold2:
- Check this story introducing the early Colab notebooks
- And this one discussing the impacts AlphaFold2 may exert on biology and on machine learning sciences.
AI/ML
Trending AI/ML Article Identified & Digested via Granola by Ramsey Elbasheer; a Machine-Driven RSS Bot
via WordPress https://ramseyelbasheer.io/2021/08/18/the-hype-on-alphafold-keeps-growing-with-this-new-preprint/


